

서 론
재료 및 방법
26 종의 벼 품종 전장유전체 분석
통계분석
GWAS 분석
결과 및 고찰
농업형질에 대한 상관관계 및 주성분 분석
26 개 벼 품종의 DNA 다형성
계통분석 및 GWAS
서 론
벼(Oryza sativa L.)는 밀, 옥수수를 비롯해 세계적으로 재배되고 있는 3 대 식량 작물이다. 120 여 국가에서 재배되고 있는 벼는 총 생산량의 92%가 아시아 지역에서 생산되며 총 소비량의 90%가 이들 지역에서 소비되고 있다. 최근 지속적으로 출산율은 감소하지만 기대 수명의 증가로 식량확보가 절실히 필요한 시점이다. 통계청 자료에 의하면 우리나라는 2013년을 기준으로 단위 면적당 쌀 생산량을 계속해서 증가 유지하였지만 2016년을 기준으로 계속해서 생산량과 재배면적은 줄고 있는 실정이고 증가하는 세계인구에 식량을 공급하기 위해 2035년까지 1억 1,600만톤의 쌀이 추가로 필요할 것으로 예상 된다(Seck et al., 2012). 우리나라의 벼 품종 육성의 시대적 변천 과정은 1960년 국제 미작 연구소(International Rice Research Institute, IRRI)가 설립되면서 활성화 되었다. 1970년 수량성을 증대시키기 위하여 개발한 ‘통일벼’ 품종이 1980년 냉해로 생산량이 크게 줄어든 사건을 계기로 다양한 농업 형질 개량과 재배법 개선 등이 이루어져 안정화가 되어갔다. 최근에는 식량 확보를 위한 작물의 수량성 증가보다는 내병성, 고식미 및 기능성 성분 증대에 관심이 높아지고 있으며 벼에 대한 신품종 개발이 지속해서 이루어지고 있다.
벼의 분자육종에서 유전체 해독을 위한 차세대 염기서열 분석(Next Generation Sequencing, NGS) 기술은 신속하면서도 저비용으로 유전자 분자 표지를 확인하는 방법으로 널리 활용되고 있다. 이 기술의 발전으로 이전에 없었던 대규모의 유전체 정보를 확보할 수 있게 되었고, 대체적 스플라이싱(alterative splicing) 유전자들과 뉴클레오타이드 서열 내 다양성, 중요 목적 유전자, 또는 광범위한 분자 표지 모티브 다형성에 있어 새로운 장을 제공하게 되었다(Garg et al., 2011; McDowell et al., 2011; Sloan et al., 2012). Genome-Wide Association Study (GWAS)는 100 만개 이상의 단일염기서열 다형성(Single nucleotide polymorphism; SNP)들을 대량의 유전체 정보를 통해서 개개인의 유전자형(Genotype)들을 결정하고, 질병이나 특정표현형과 같은 다양한 형질에서 유전자형-표현형의 특이적 관련성을 나타내는 돌연변이를 발굴하는 분석이다(Kruglyak, 1999). 이 전 연구에서는 세계적으로 밀의 중요한 질병인 얼룩반점병저항성(Spot Blotch, SB)에 이전에 확인된 유전자 3 개와 그 외의 유전자 6 개를 추가 식별하여 관련성을 검증했을 뿐 아니라(Ayana et al., 2018), 또 다른 연구에서는 여러 형질들의 주성분 분석을 통하여 녹두 유전자원 집단에서 GWAS분석으로 녹두 종자 색에 연관된 유의미한 유전자좌를 동정하는 등 GWAS 분석을 이용한 연구가 매우 활발히 이루어지고 있다(Daovongdeuan et al., 2017). 반면 유전체 정보를 수집함에서 서로 다른 영역에 위치한 염색체가 둘 이상의 양적 형질이나 복잡한 질병을 수반하는 경향이 많아 다수의 형질과 연관이 있는 공통 유전자를 조사하는 것이 최근 연구 추세이다(Gao et al., 2014).
본 연구에서는 26 종의 벼 품종에 대한 대량의 유전체 정보에서 10 가지 농업 형질과 관련한 GWAS 분석을 실시하였으며 각 유용 형질과 관련한 유전자좌를 탐색하고자 하였다.
재료 및 방법
26 종의 벼 품종 전장유전체 분석
26 종 벼 품종의 유전형 정보는 Whole Genome Sequencing, Resequencing, RNASeq의 키워드로 검색하여 Illumina Hisec4000 플랫폼으로 resequencing된 데이터를 다운받아 사용하였고(NABIC, http://nabic.rda.go.kr/), 벼의 표준 유전체 염기서열에는 IRSGP 1.0 (International Rice Genome Sequencing Project)를 이용하였다. Samtools, GATK tool 프로그램을 사용하여 SNP 확보와, 양질의 DNA 단편을 확보하기 위한 불필요한 염기서열(Genome Quality score < 21 , Phred Quailty Score < 21 , missing rate > 0.4)제거를 수행 하였다(McLaren et al., 2016). 해당 품종에 대한 10 가지 농업 형질(출수기, 초장, 주당수수, 수당립수, 등숙률, 천립중, 도열병저항성, 흰잎마름병저항성, 줄무늬잎마름병저항성, 벼멸구저항성) 정보는 농사로를 통해 수집하였다(농사로, http://www.nongsaro.go.kr/).
통계분석
10 가지 농업형질의 Principal Component Analysis (PCA)분석은 R studio에서 R package (Library)를 사용하여 분석하였으며, 상관관계는 Pearson’s Correlation Coefficient (PCC)를 통해 평가하였다(Pearson, 1896). 26 종 벼 품종의 계통분기지도는 SNPhylo 프로그램을 사용하여 도식화하였다(Lee et al., 2014).
GWAS 분석
벼의 표준 유전체에서 염기의 위치를 기반으로 SNPs와 indels를 Ensembl Variant Effect Predictor (VEP; https://asia.ensembl.org/info/docs/tools/vep/index.html)를 이용하여 유전자에 대한 정보를 특성화하였다. 분석된 4 개의 주성분(PC1, PC2, PC3, PC4)에 대한 GWAS 분석은 Plink 프로그램(http://zzz.bwh.harvard.edu/plink/)을 사용하여 수행하였고 후보 유전자들의 정보는 rap-db 사이트(https://rapdb.dna.affrc.go.jp/)에서 수집하였고 후보유전자들이 위치한 자리의 QTL정보는 3000 개의 벼 품종의 GWAS결과 데이터 snp-seek 사이트(https://snp-seek.irri.org/)에서 수집 하였다.
결과 및 고찰
농업형질에 대한 상관관계 및 주성분 분석
GWAS 분석은 일반적으로 Generalized Linear Model (GLM) 또는 Analysis of Variance (ANOVA)와 같은 통계적 방법을 사용하여 조사된 형질의 정규분포를 기초로 분석을 한다(Bush et al., 2012). 따라서 수집한 조사결과의 정규분포에 대한 유의성을 검정하기 위하여 10가지 농업 형질(출수기, 초장, 주당수수, 수당립수, 등숙률, 천립중, 도열병저항성, 흰잎마름병저항성, 줄무늬잎마름병저항성, 벼멸구저항성)에 대한 통계분석을 실시하였다(Fig. 1A). 그 결과, 10 가지의 농업형질에 대한 수집된 조사결과는 정규분포와 통계적으로 유의한 차이(p < 0.05)를 보이며 특정 수치에 편중되어 있음을 나타내었다. 형질들 사이의 상관관계 분석에서 수당립수와 등숙률 사이의 비교적 낮은 음의 상관계수(r = -0.64)는 통계적 유의성(p < 0.05)을 보였으며 수당립수가 많아질수록 등숙률이 낮아진다는 것을 반영한다. 또한 출수일과 벼멸구저항성 사이에서 통계적으로 유의한 음의 상관관계(r = -0.63)를 보였다. 이는 상대적으로 출수일이 빠른 벼 품종의 경우 벼멸구저항성에 상대적으로 더 취약함을 나타낸다. 이외에 다른 형질들 사이에서는 상관관계의 통계적 유의성을 관찰할 수 없었다.
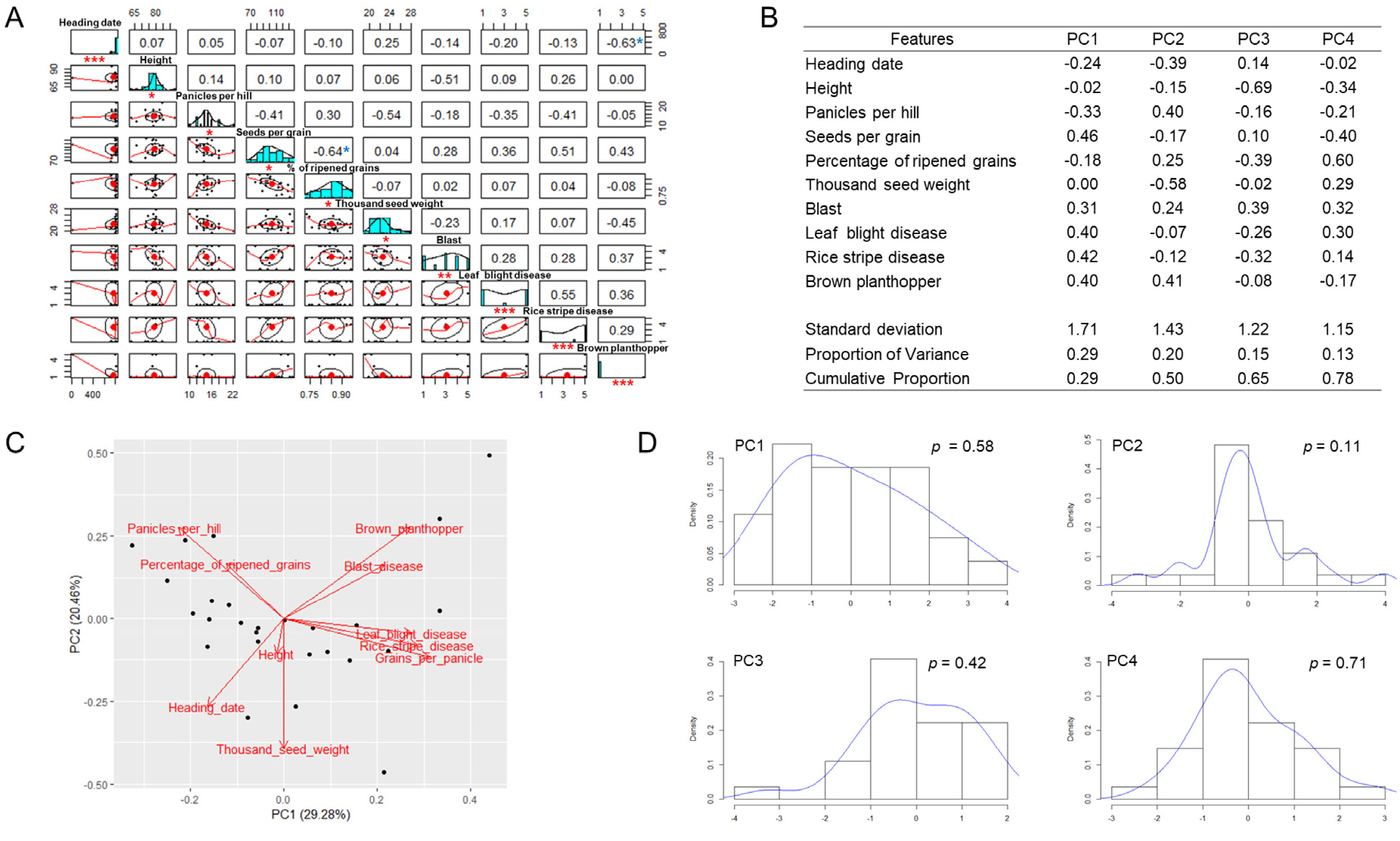
Fig. 1
Principal component analysis (PCA) of different agronomic traits. A: The distribution and correlation of ten traits (heading date, height, panicles per hill, seeds per grain, percentage of ripened grains, thousand seed weight, blast, leaf blight disease, rice stripe disease, and brown planthopper). The number indicates Pearson’s correlation coefficient. Star indicates significant differences determined using a Student’s t-test with n-2 degrees of freedom (blue stars) or Shapiro-Wilk test (red stars). *p < 0.05; **p < 0.01; ***p < 0.001. B: PCs of the phenotype scores of the ten traits. C: Loading plot between PC1 and PC2. The percentage in parentheses represents the proportion of variance. D: Bar plots of PCs. The Shapiro-Wilk test was used to determine significant difference (p).
Yano et al. (2019)은 벼에서 농업형질을 조절하는 유전자를 확인하기 위하여 특정 통계 수치에 편향된 자료를 토대로 PCA 분석을 수행하여 정규분포의 주성분 값을 이용하여 GWAS 분석을 실시하였다(Yano et al., 2019). 따라서 이 연구에서도 특정 통계 수치에 편중된 10 개의 농업형질에 관한 PCA 분석을 실시하였다(Fig. 2). 그 결과, PC1에서 PC4까지의 0.78의 70% 이상의 누적된 분산과 각 주성분(PC) 값에서 상대적으로 높은 수치를 보이는 형질을 확인하였으며(Fig. 1B). PC1은 수당립수(0.46)에서 가장 높은 주성분 값을 보였으며 흰잎마름병저항성(0.42), 줄무늬잎마름병저항성(0.44), 벼멸구저항성(0.40)에서 상대적으로 높은 수치를 나타내었다. 이 결과는 PC1의 주성분 값이 26 개 벼 품종에서 수당립수와 같은 높은 수치를 보이는 형질을 설명하기 적합하다는 것을 제시한다. PC2는 벼멸구저항성(0.41)과 주당수수(0.40)에서 비교적 높은 수치를 보였으며 천립중(-0.58)에서 낮은 음의 수치를 보였다. PC3는 신장에서 -0.69의 낮은 수치를 보였으며 PC4는 등숙률(0.60)에서 상대적으로 높은 수치를 나타냈다. 가장 높은 분산을 보이는 주성분 PC1 (29.28%)과 PC2 (20.46%)에서 각 형질의 상관관계를 관찰하였다(Fig. 1C). 그 결과, 2 개의 주성분을 통하여 분석된 품종 사이에서 흰잎마름병저항성, 줄무늬잎마름병저항성, 수당립수의 형질이 유사함을 관찰하였다. PC1에서 PC4까지의 각 품종에 대한 주성분 값을 토대로 정규분포 유의성을 검정하였다(Fig. 1D). 그 결과, 4 개의 주성분은 정규분포와 통계적으로 유의한 차이가 없음(p > 0.05)을 나타내었으며 이를 이용한 GWAS 분석의 가능성을 제시하였다. 이 전 연구에서 Cubry et al. (2020)은 아프리카 벼인 Oryza glaberrima를 개화시기와 구조적 특성 및 Rice yellow mottle virus를 포함한 형질들의 PCA분석을 통하여 나온 주성분 값을 이용하여 GWAS를 실시하였고 결과로서 QTL 및 후보유전자인 Rymv1, Short Panicle 1 (SP1)과 구조적 특성에 관련한 장일 조건에서 벼 출수와 곡물생산 증가를 야기하는 Ghd7.1과 APO1, OsMADS1 등 관련 형질에 대한 유전자를 확인하였다(Cubry et al., 2020; Yan et al., 2013).
26 개 벼 품종의 DNA 다형성
26 개 벼 품종 사이에서 발생한 유전적 변이를 관찰하기 위하여 SNPs와 indels와 관련한 DNA 다형성을 분석하였다(Fig. 2). 그 결과, 2,527,855 SNPs와 381,686 indels를 발견하였고 1번 염색체에서 가장 높은 수의 SNPs (261,071)와 indels (42,412)를 나타났으며 11번 염색체, 6번 염색체 2번 염색체 등의 순으로 나타났다. 반면에 9번 염색체에서 가장 낮은 수의 SNPs (151,274)와 indels (21,943)가 관찰되었다(Fig. 2A). 이는 26 개의 국내 벼 품종 사이에서 상대적으로 차이가 나타나는 염색체의 유전적 변이 양상을 제시한다.
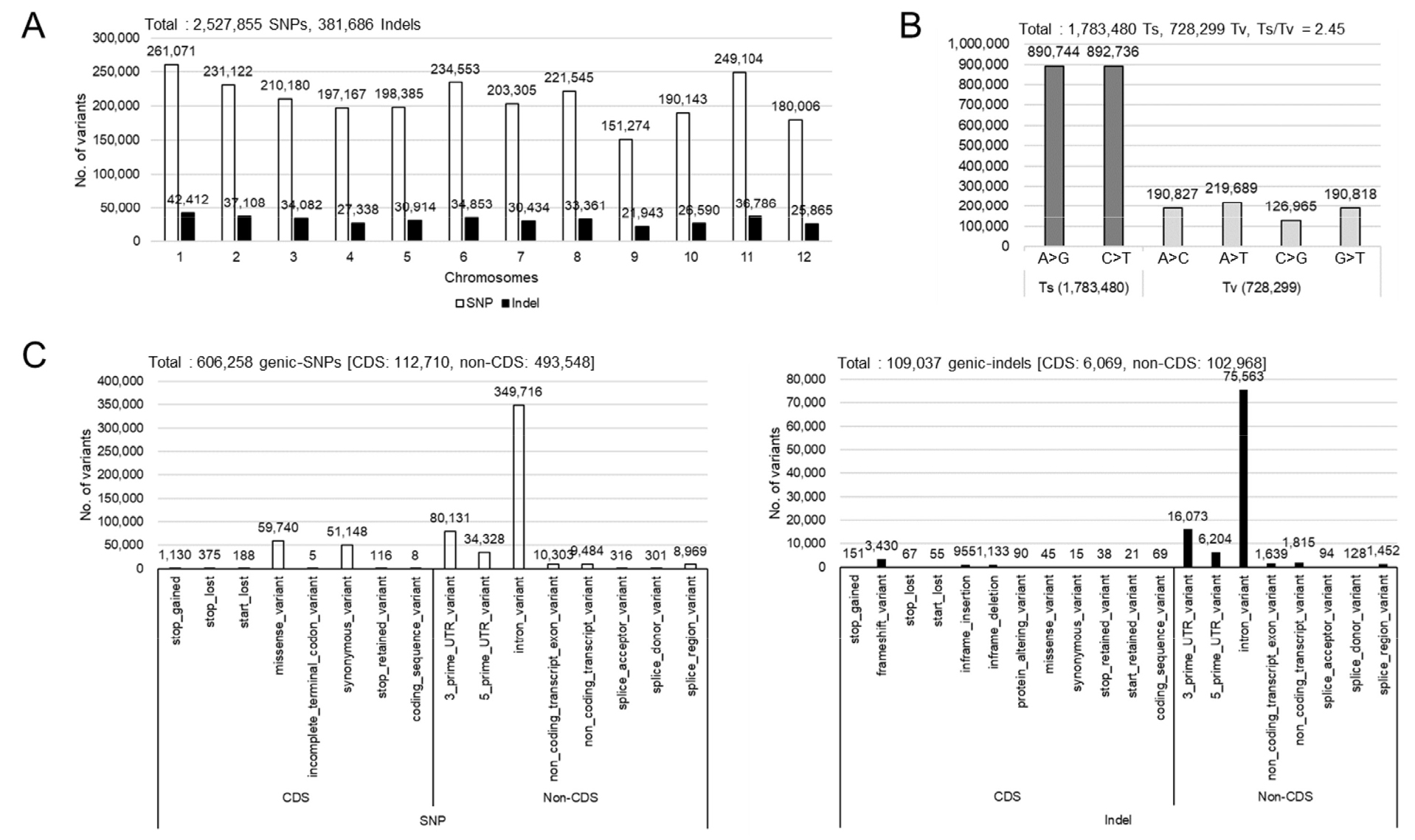
Guo et al. (2017)은 연구를 통해 전환(Transversion; Tv)이 전이(Transition; Ts)보다 DNA의 분자 구조에서 너비와 기능을 변경시켜 전사 인자 결합에 더 큰 영향을 준다는 것을 확인하였다(Guo et al., 2017). 따라서 국내 벼 품종 사이에서 발생한 SNPs 중에서 발생한 전이와 전환의 발생빈도를 관찰하였다(Fig. 2B). 퓨린과 피리미딘 사이에서 염기의 구조 변화에 영향을 주지 않는 전이(Transition; Ts)의 경우 1,783,480 SNPs, 구조에 변화를 나타내는 전환(Transversion; Tv)의 경우 728,299 SNPs로 나타났다. 전이(Transition; Ts)의 경우 A와 G, C와 T사이에서 발생한 SNPs의 수는 큰 차이를 보이지 않았다. 전환(Transversion; Tv)의 경우 A와 T 사이에서 가장 높은 수의 변이(219,689 SNPs)가 관찰되었으며 C와 G 사이에서 가장 낮은 수의 변이(126,965 SNPs)가 나타났다. 26 개 벼 품종 사이에서 나타나는 전환에 대한 전이의 비율(Ts/Tv ratio)는 2.45로 관찰되었다. Lekklar 등은 태국의 벼 30품종에서 2.4에서 2.42의 Ts/Tv ratio를 관찰하였으며 이러한 결과는 국내 벼 품종 사이에서 보이는 비율과 유사한 수치였다(Lekklar et al,. 2019).
Guo et al. (2017)은 연구를 통해 전환(Transversion; Tv)이 전이(Transition; Ts)보다 DNA의 분자 구조에서 너비와 기능을 변경시켜 전사 인자 결합에 더 큰 영향을 준다는 것을 확인하였다(Guo et al., 2017). 따라서 국내 벼 품종 사이에서 발생한 SNPs 중에서 발생한 전이와 전환의 발생빈도를 관찰하였다(Fig. 2B). 퓨린과 피리미딘 사이에서 염기의 구조 변화에 영향을 주지 않는 전이(Transition; Ts)의 경우 1,783,480 SNPs, 구조에 변화를 나타내는 전환(Transversion; Tv)의 경우 728,299 SNPs로 나타났다. 전이(Transition; Ts)의 경우 A와 G, C와 T사이에서 발생한 SNPs의 수는 큰 차이를 보이지 않았다. 전환(Transversion; Tv)의 경우 A와 T 사이에서 가장 높은 수의 변이(219,689 SNPs)가 관찰되었으며 C와 G 사이에서 가장 낮은 수의 변이(126,965 SNPs)가 나타났다. 26 개 벼 품종 사이에서 나타나는 전환에 대한 전이의 비율(Ts/Tv ratio)는 2.45로 관찰되었다. Lekklar 등은 태국의 벼 30품종에서 2.4에서 2.42의 Ts/Tv ratio를 관찰하였으며 이러한 결과는 국내 벼 품종 사이에서 보이는 비율과 유사한 수치였다(Lekklar et al,. 2019).
유전자의 기능 변화에 큰 영향을 줄 수 있는 변이를 탐색하기 위하여 유전자의 염색체에 대한 물리적 위치를 기초로 하여 SNPs와 indels를 분류하였다(Fig. 2C). 전체 606,258 SNPs와 109,037 indels가 벼의 유전자 부위에서 발견되었다. 유전자 부위의 변이 중에서 coding sequence (CDS)에서는 112,710 SNPs와 6,069 indels가 나타났으며 CDS가 아닌 지역에서 493,548 SNPs와 102,968 indels가 관찰되었다. CDS 지역에서 발생한 SNPs 중에서 missense SNPs (59,740)와 synonymous SNPs (51,148)가 주로 분포하였으며 CDS 이외의 intron 지역에서 대부분의 SNPs (349,716)가 발생하였다. indels의 경우 CDS 내에서 frameshift 변이(3,430 indels)가 다수 관찰되었으며 SNPs와 유사하게 intron 부위(75,563 indels)에서 주로 변이가 나타났다.
계통분석 및 GWAS
26 개 벼 품종 사이의 DNA 다형성과 관련한 유연관계를 분석하기 위하여 계통지도를 작성하였다(Fig. 3A). 그 결과, 5 개 그룹의 유연관계가 상당히 밀접되어 있는 품종들을 확인하였다. 예를 들어 Group I은 유남, 신동지, 화영, 호품, Group II는 조평, 녹양, 다산, 한아름, 삼강, 밀양-23, 장성-1, 단양-38, Group III은 팔공, 밀양99h, 호평, Group IV는 추청, 낙동, 화성, Group V는 오대벼와 진부벼 품종이 포함되었다. Group I의 화영벼는 양질, 내병 다수성 품종을 육성하고자 흰잎마름병저항성에 강하면서 단간인 쥬케이 830과 도열병 저항성 및 줄무늬잎마름병저항성에 강한 YR4811Acp8을 인공 교배시켜 잡종세대를 양성하였다. 신동진벼는 고품질 양질 다수성 품종육종을 목적으로 양질이고 내병성인 화영벼와 YR13604Acp 22계통을 인공 교배하여 다수성 계통을 선발한 후 신동진벼라 명명하였다. 이는 계통분기지도의 화영벼와 신동진벼의 밀접한 유연관계를 뒷받침하였다. Group II의 한아름벼는 내도복 복합내병충성을 갖춘 초 다수성 품종을 육성할 목적으로 내병충성 초 다수성인 밀양벼에 초형이 양호하고 흰잎마름병저항성이 강하며, 초 다수성인 수원405호를 인공 교배하였다. 또한 삼강벼는 여러 가지 재해에 견디는 양질 다수성 신품종을 육성할 목적으로 내병충 다수성인 밀양벼를 모본으로 하였다. 장성벼는 벼멸구저항성에 강하고 양질 다수성인 품종을 육성하기 위하여 밀양29호, 밀양30호의 F1을 복교잡한 후 계통육종법에 의하여 육성 선발 하였다. 벼 품종에서 한아름벼, 삼강, 밀양-23, 장성의 경우 모두 밀양벼를 부 또는 모로써 교배 육종한 품종이며 이는 계통분기지도와 일치한 결과를 보였다.
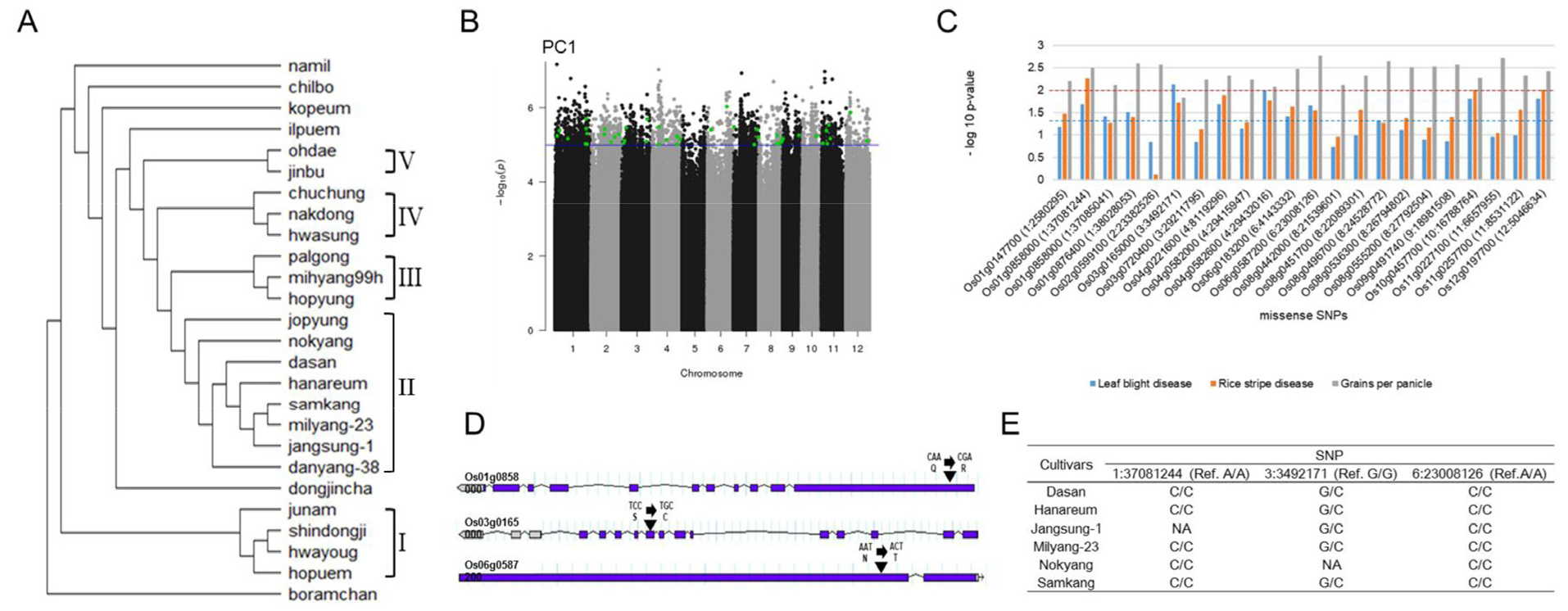
Fig. 3
The phylogeny of Korean rice cultivars and genome-wide associate study (GWAS) using PC1. A: Phylogenetic tree constructed using the neighbor-joining method. B: Manhattan plot of single-nucleotide polymorphisms (SNPs) on chromosomes. Green dots indicate missense SNPs. The blue line represents a significant level (p < 0.00001). C: Phenotypic differences between plants with and without significant SNPs. Bar color represents different traits. The line indicates significant differences determined using the Mann–Whitney U test (blue color, p < 0.05; red color, p < 0.01). D: SNPs detected in the three candidate genes (Os01g0858000, Os03g0165000, and Os06g0587200). E: Korean rice cultivars with the three candidate SNPs.
4 개의 주성분(PC1-PC4)이 반영하는 각각의 형질과 밀접한 관련이 있는 SNPs를 탐색하기 위하여 GWAS 분석을 실시하였다(Fig. 3B). 그 결과, PC1의 주성분을 이용하였을 때 2,168 SNPs이 -log10(p)에서 5이상의 높은 통계적 유의성을 보였으며 그 중 45 개의 SNPs이 아미노산의 변이를 가져오는 missense variant였다. 반면에 PC2, PC3, PC4에서는 아미노산의 변화에 영향을 미치는 SNPs 중에서 임계값(-log10(p) = 5)을 만족하는 변이를 관찰하지 못하였다. PC1의 GWAS 분석을 통하여 분석된 45 개의 missense SNPs의 변이가 26 개 품종에 형태적 영향을 미치는지를 확인하기 위하여 PC1에서 높은 주성분 값을 보인 3가지 형질(흰잎마름병저항성, 줄무늬잎마름병저항성, 수당립수)에서 SNPs의 발생 유무에 따라 두 그룹으로 나눈 후 통계적으로 유의한 차이를 관찰하였다(Fig. 3C). 통계적 분석을 위하여 45 개의 missense SNPs 중에서 5 개 이상의 식물에서 변이를 관찰할 수 없었던 23 개의 SNPs는 제외하였다. 그 결과, 22 개의 missense SNPs 중 Os03g0165000 (DNA topoisomerase 3 protein)은 흰잎마름병저항성, Os01g0858000 (AT hook motif family protein)은 줄무늬잎마름병, Os06g0587200 (Protein kinase, catalytic domain domain containing protein)은 주당립수의 형질에서 변이를 보이는 식물의 형태적 특징의 차이가 SNPs을 가지지 않은 식물과 비교하여 상대적으로 높은 유의성을 나타내었다. 상대적으로 높은 유의성을 보인 3 개의 유전자들의 SNP와 아미노산 변형을 관찰한 결과, Os03g0165000에서 A에서 G의 변이가 Glutamine (Q)에서 Arginine (R)으로의 아미노산 변형에 영향을 주었으며 Os03g0165000은 TCC (Serine; S)에서 TGC (Cysteine; C), Os06g0587200은 AAT (Asparagine; N)에서 ACT (Threonine; T)로의 변화를 나타내었다(Fig. 3D). SNPs의 발생에 따른 형태적 특징 비교에서 상대적으로 높은 유의성을 보인 3 개의 SNPs는 6 개의 품종에서 관찰되었으며 모두 계통지도에서 Group II에 속하였다(Fig. 3E). 이러한 결과는 흰잎마름병, 줄무늬잎마름병, 수당립수에 관련한 3 개의 SNPs가 Group II의 대부분 품종들이 공통적으로 나타내는 복합내병충성, 흰잎마름병저항성, 다수성과 같은 형질에 영향을 미쳤음을 제시한다.
후보 유전자들이 기존에 알려진 QTL 지역에 속해 있는지 여부를 판단하기 위하여 각 유전자들의 정보를 데이터베이스를 통해 검색하였다(https://snp-seek.irri.org/). 그 결과, Os01g0858000의 위치에서는 식물의 무게(qPHT-1), 유아출현(Dth1.1) 등 줄무늬잎마름병 저항성과는 관련하지 않은 QTL이 확인 되었고 Os03g0165000에서도 개화시기(QTAROqtl-861), 식미(qTA3) 등 흰잎마름병 저항성과는 관련 없는 QTL이 확인되었다. 반면 Os06g0587200의 경우 종자의 길이와 폭(qtl-5), 천립중(Tgw6.1), 종자 폭(Gw6)에 관련한 QTL에 속해 있음을 관찰하였고 이는 해당 관련 형질인 수당립수와 연관이 있음을 확인 하였다. 또한, 3 개의 후보 유전자들 내에서 발생한 SNP의 존재 여부를 26 개 품종에서 확인한 결과, 6 개 품종 내에서 동일한 SNP의 발생 빈도를 관찰하였다. 6 개 품종의 계통 유연관계는 매우 밀접하였으며 이는 복합내병성 저항성을 가진 밀양벼에서 기원하였다. 따라서 본 연구에서 나타난 후보유전자 중 2 개의 내병성 관련 유전자와 1 개의 종자 수 관련 유전자가 다른 계통들과 비교해 밀양벼 관련 계통들의 유용 형질을 조절하는 중요 유전자임을 제시한다. 이러한 GWAS의 접근 방법은 복잡한 형질과 관련한 후보유전자의 효율적 선발을 위하여 활용될 수 있을 것이다.
